
One thing I understand, despite my little experience, is that sequencing DNA is relatively simple. After all, it is enough to fragment the DNA or cDNA to be sequenced, build a library with the fragments obtained and "read" the sequence of the individual fragments, thus obtaining different reads. After sequencing we proceed with the assembly of the reads obtained in order to reconstruct the entire sequence of the sequenced DNA or cDNA.
But before proceeding with the assembly it is necessary to do some cleaning. In fact, the reads obtained are not really suitable for assembly, it is in fact necessary to remove the adapters (if present) and filter the reads on the basis of specific quality criteria that are summarized by a statistical value defined as Phred. In general, Phred values greater than 30 are considered acceptable, as can be seen from the table below.

The Phred is given by the following relation:
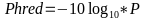
where P is the probability of error in calling the nitrogenous bases during sequencing.

In general, there are three approaches that can be used for cleaning reads:
- Fixed length trimming. It is based on the removal of the 3 ′ portions of the sequences since usually in the terminal part the reads have lower quality due to the greater tendency to accumulate errors in calling the nitrogenous bases during sequencing.
- Adaptative trimming, Excludes reads that have a length and quality less than the user-defined threshold values.
- Sliding-windows trimming. In this case, the quality of the reads is evaluated “by windows”, ie for portions 1/10 of the total length of the read. Therefore, if a window with a quality value lower than the threshold value defined by the user is encountered, the read is cut and if it respects a minimum length value, also defined by the user, it is kept otherwise it is discarded.
But let's talk a little about codes. There are several bioinformatics tools for cleaning reads but I generally use Scythe, to remove the adapters and Sickle to filter the reads, which is based on the principle of sliding windows trimming. In addition, to better evaluate the success of the cleaning you can use a software that allows you to view the quality of the reads called FASTQC.
To find out more about these programs, I suggest you consult the links I have reported below, at the end of the article.
For those interested, I also recorded a quick tutorial, reported below, on cleaning some raw sequencing data downloaded from NCBI.
Thanks for reading. As usual, I ask you to leave a comment or a "like" and if you don't want to miss the next articles I suggest you subscribe to the blog or follow me on Instagram.
Bye and see you soon.
Sources:
https://github.com/najoshi/sickle